Tutorial
AIVE Tutorial
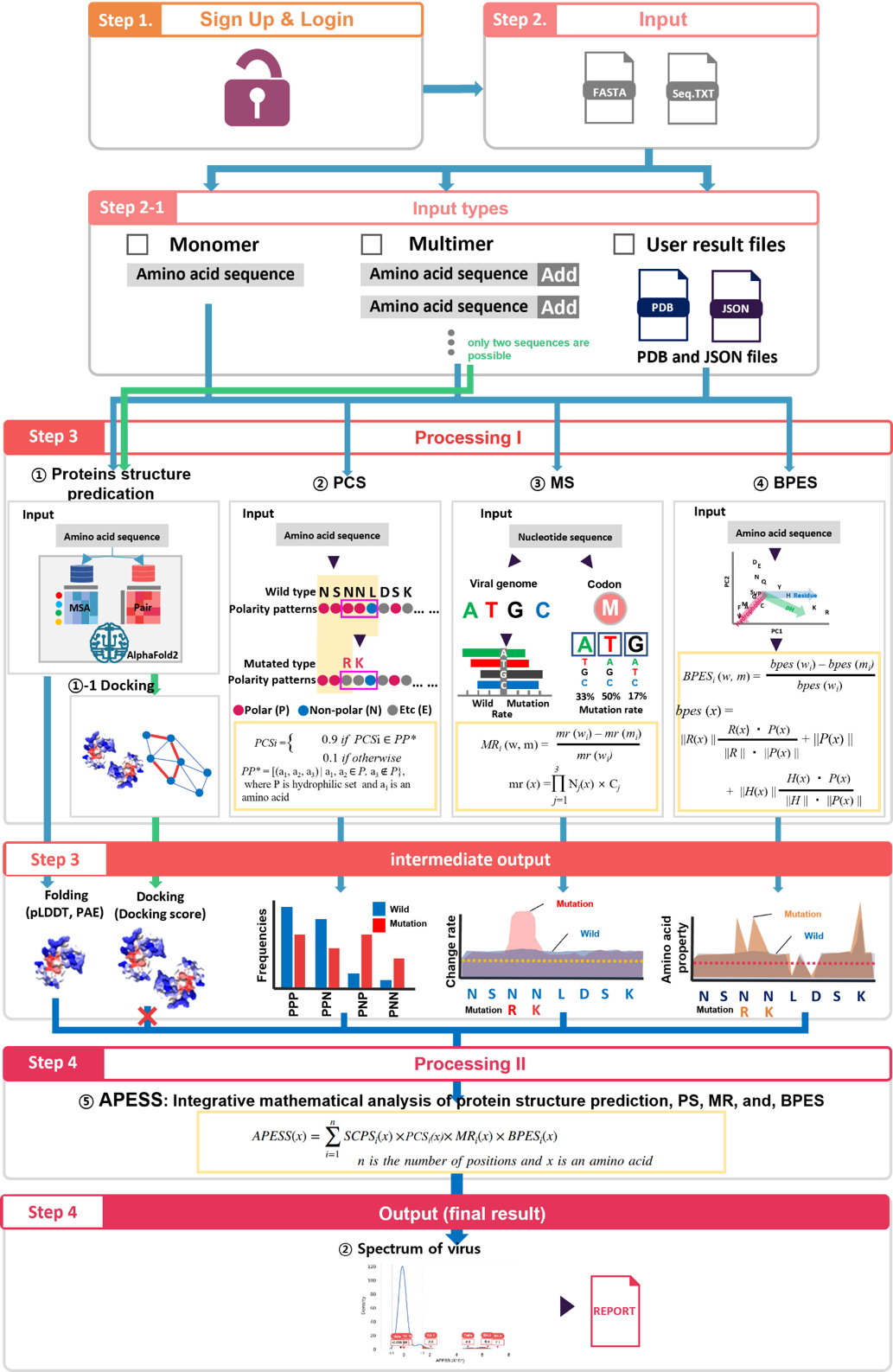
(1) AIVE can be used to evaluate :
(2) The input range of analysis for AIVE is as follows :
- - Monomer (single amino acid sequence) - Multimer (two or more amino acid sequences, when analyzing docking score, only two sequences are possible)
- - Monomer (single amino acid sequence) - Multimer (two or more amino acid sequences, when analyzing docking score, only two sequences are possible) - PDB and JSON files that users possess can be directly submitted
Sign up & Login
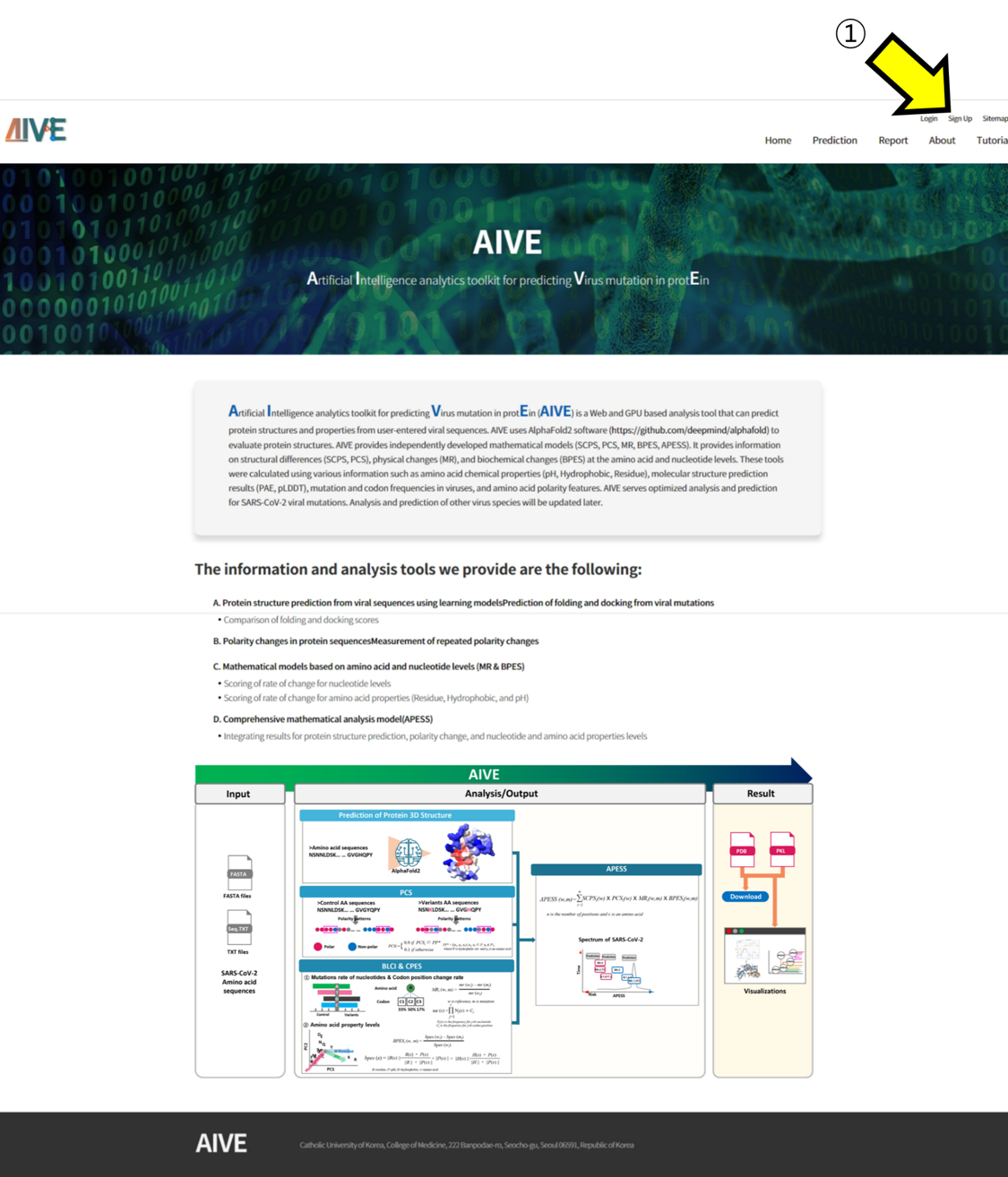
- ① Click on the "Sign Up" option to proceed with the account creation.
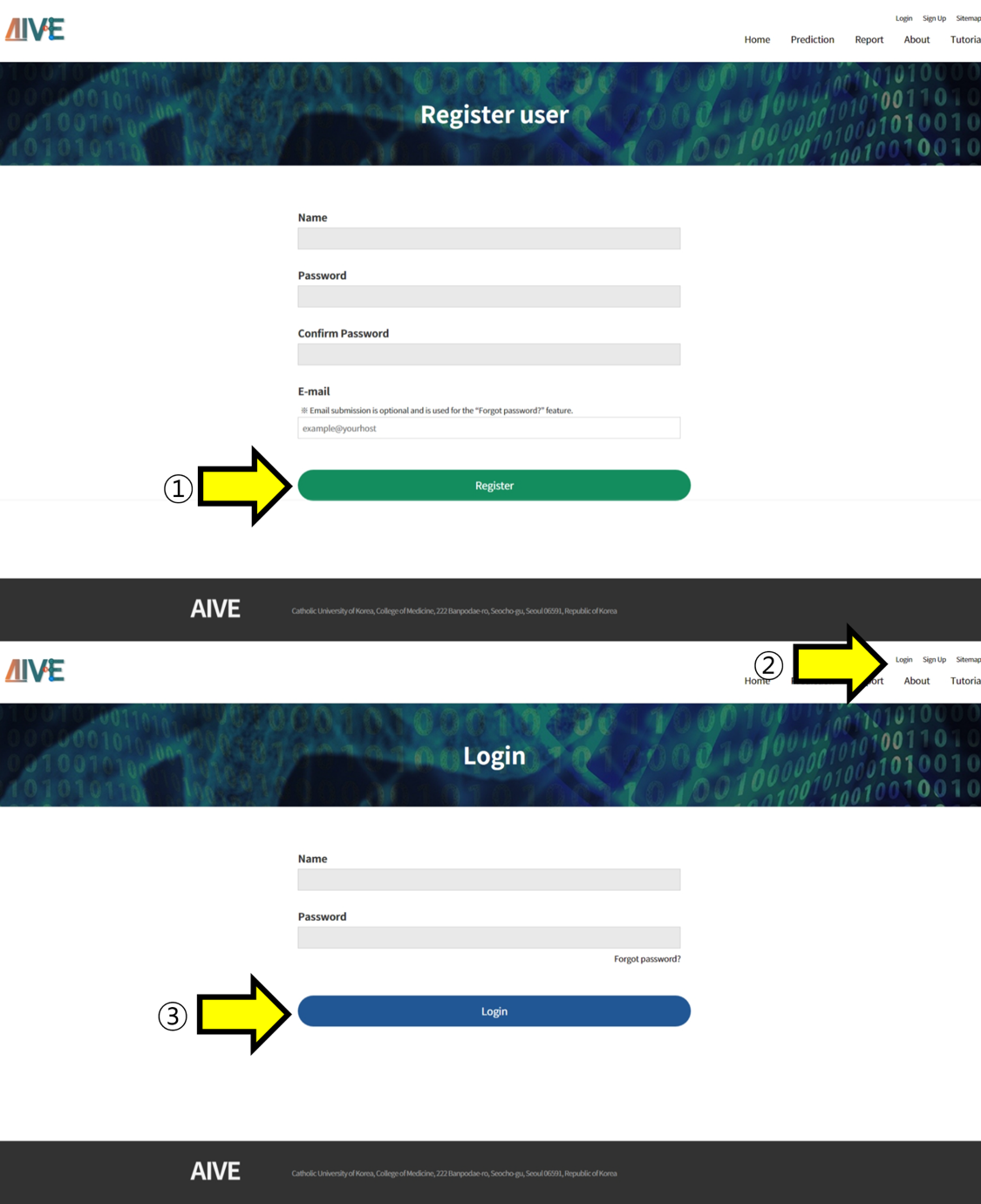
- ① Enter the username and password. To use the "Forgot password?" feature, enter your email and proceed with account creation. ②, ③ Access the "Login" section, enter the account information, and log in.
Demo
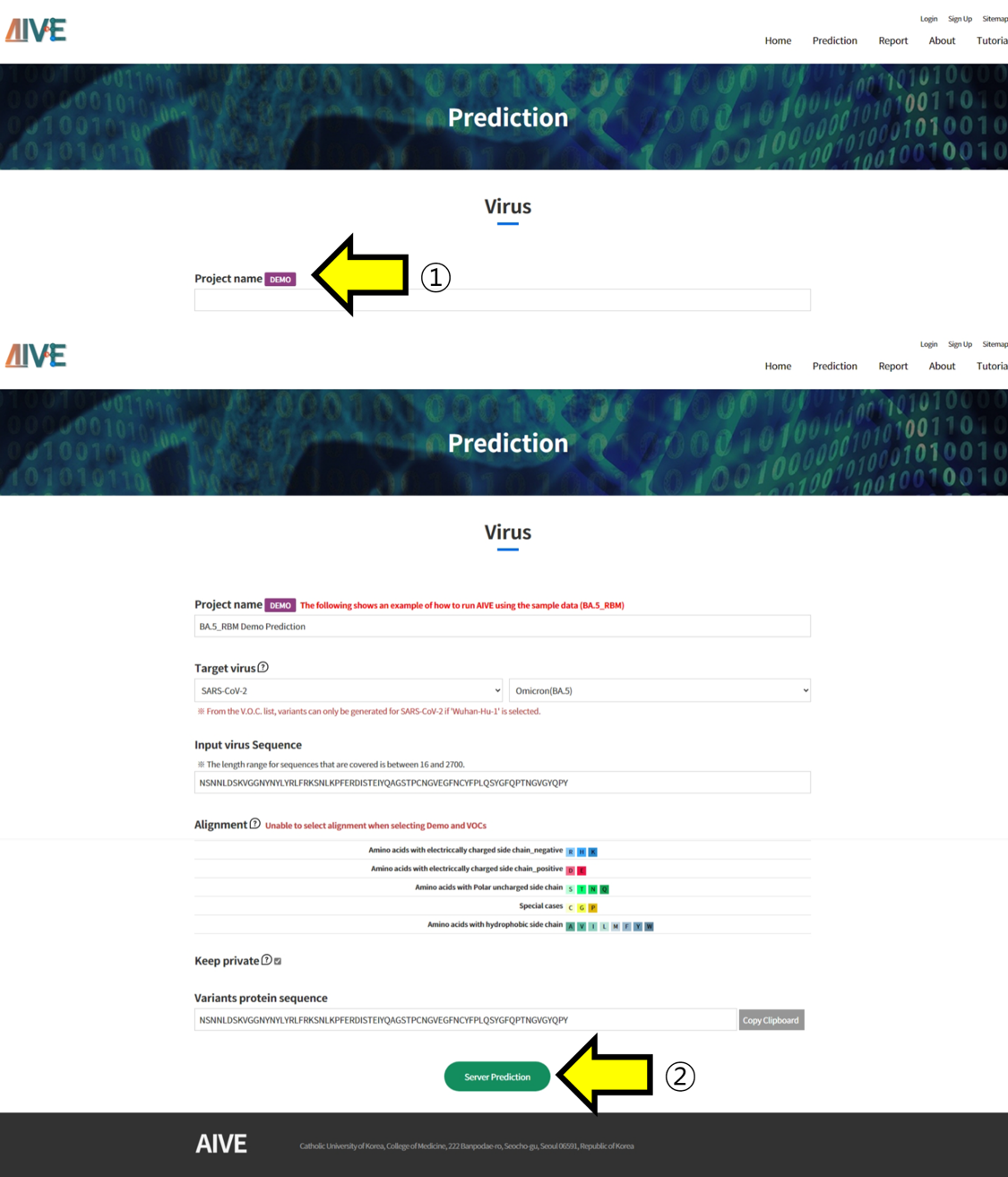
- ① Clicking on the Demo button will automatically select the RBM [S:437-508] region of the SARS-CoV-2 BA.5 variant. ② To predict and analyze the structure of the sequence generated by the Demo function, click the "Server Prediction" button to submit the task.
Predict VOC structure
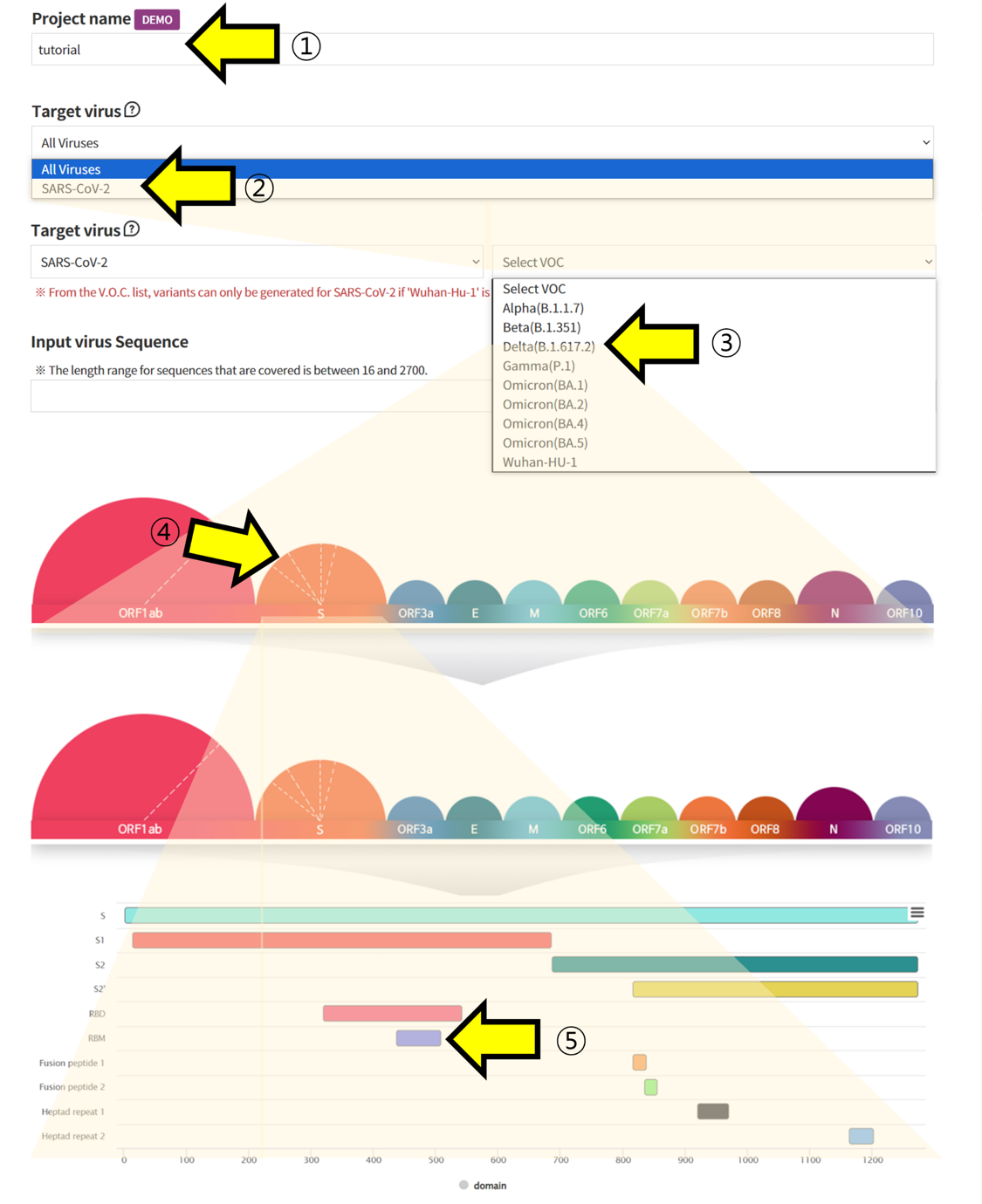
- ① Write the Project name for task categorization. ② Select SARS-CoV-2 in the Target Virus section to retrieve information on Coronaviruses. ③, ④, ⑤ Choose the VOC and regions you want to analyze to retrieve the corresponding Amino Acid sequence information.
Submit
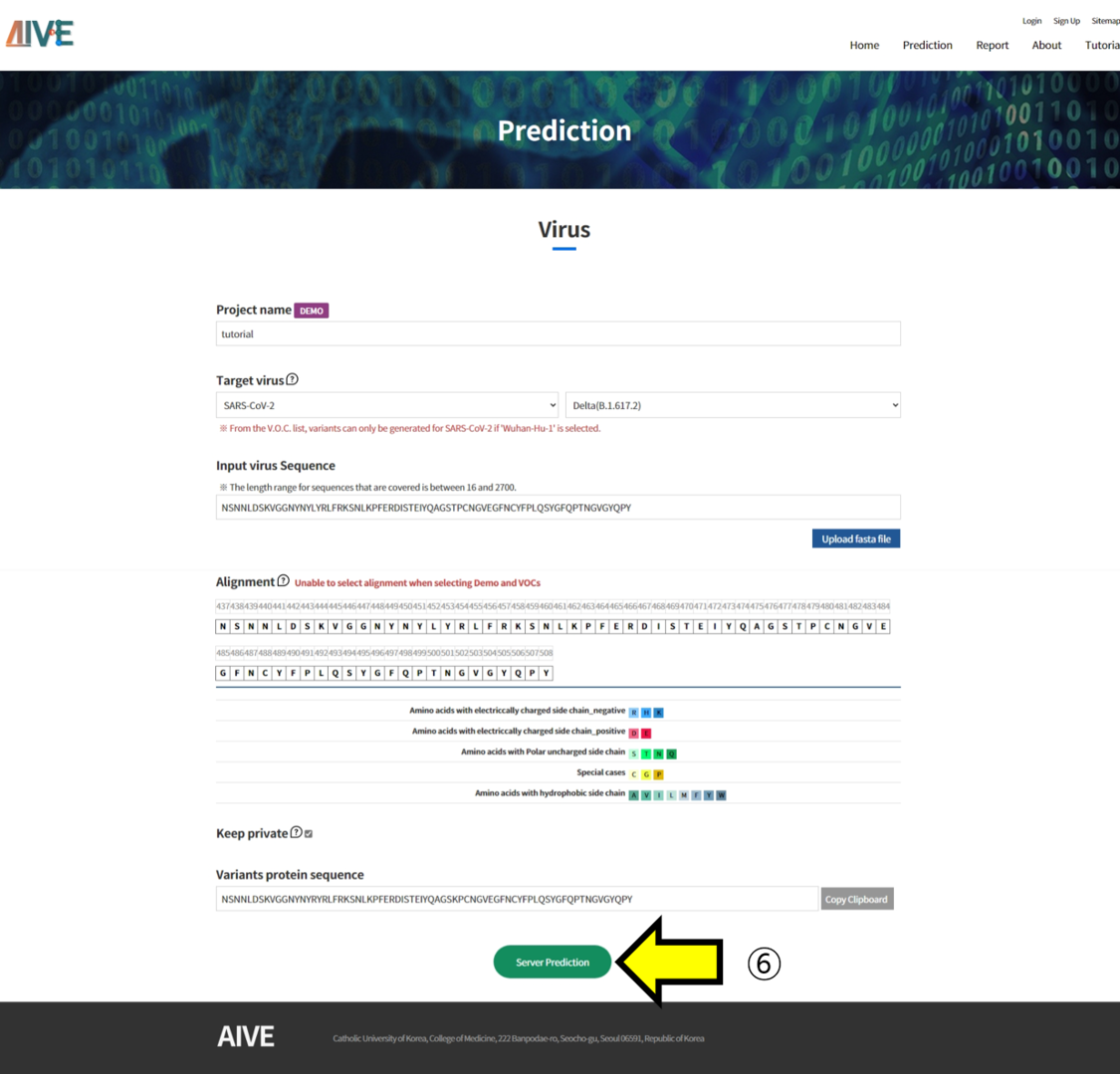
- ⑥ Click the "Server Prediction" button to start the prediction and analysis of the Project
List
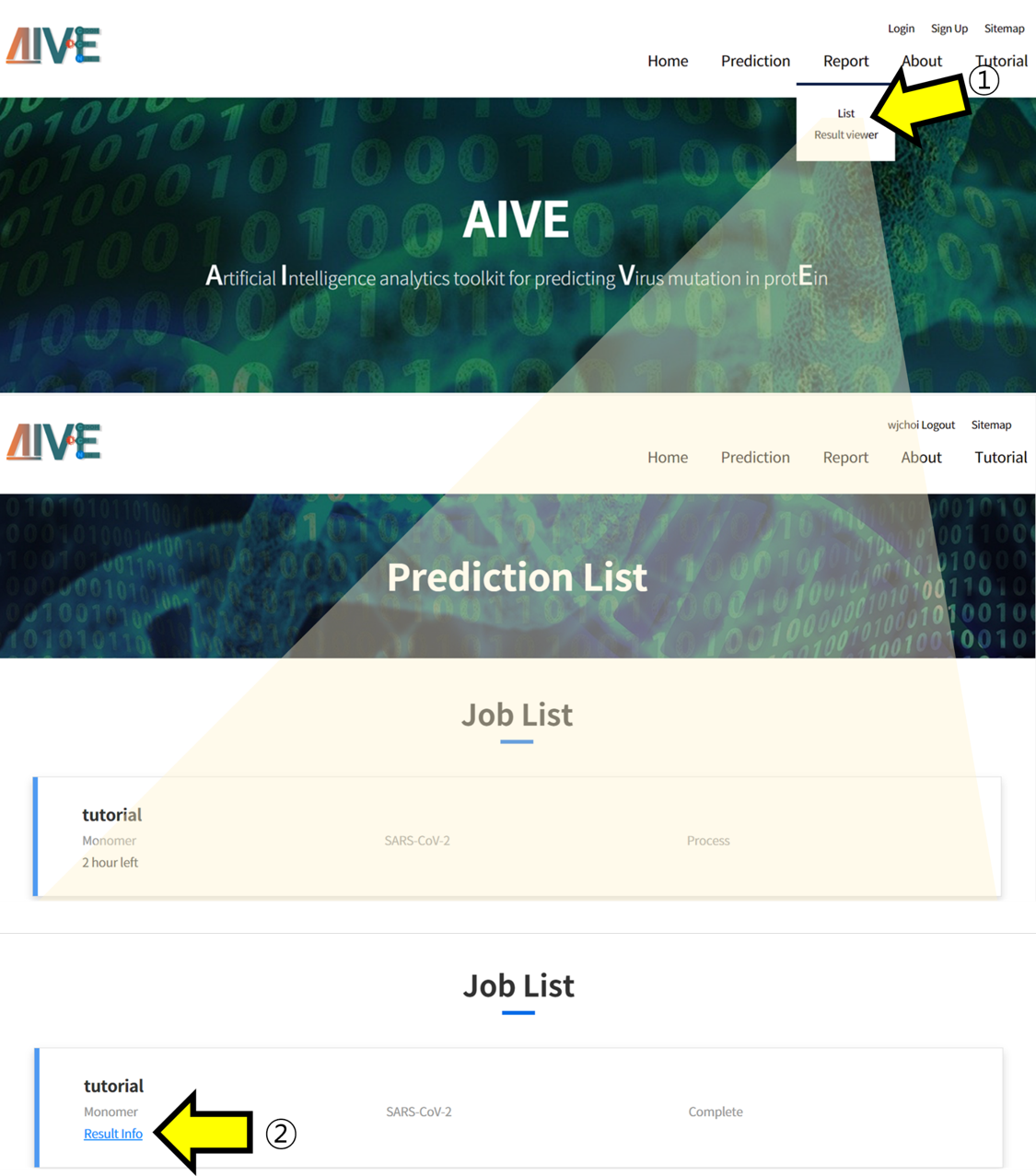
- ① Click the "Report menu” and then select "List" from the submenu to check Projects. ② You can check the analysis results by clicking on the "Result info" section for completed tasks.
Generate SARS-CoV-2 mutated sequence
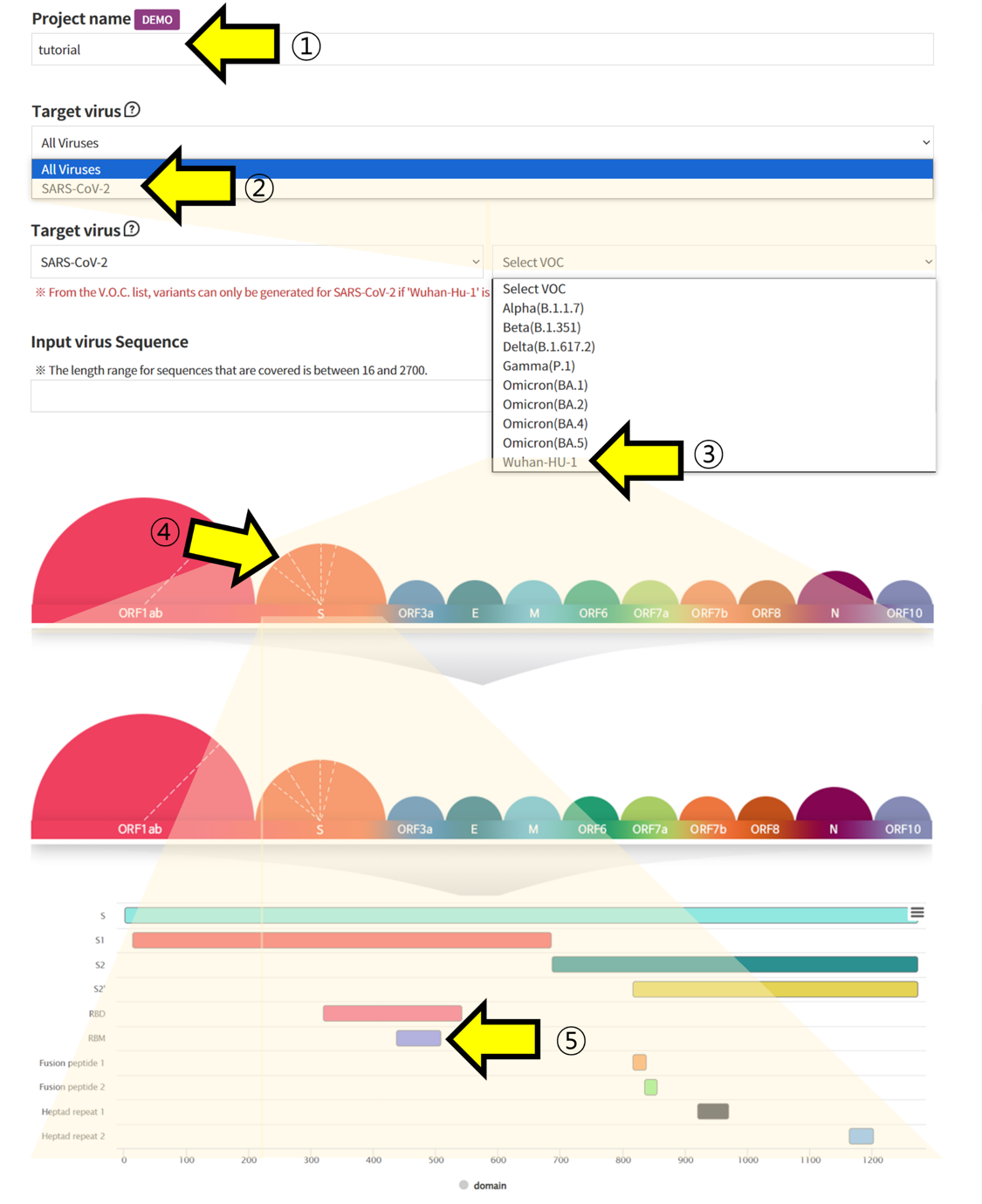
- ① Specify a Project name to categorize the submitted task ②, ③, ④, ⑤ Choose Wuhan-HU-1 sequence of SARS-CoV-2 and retrieve the sequence of the region you want to verify.
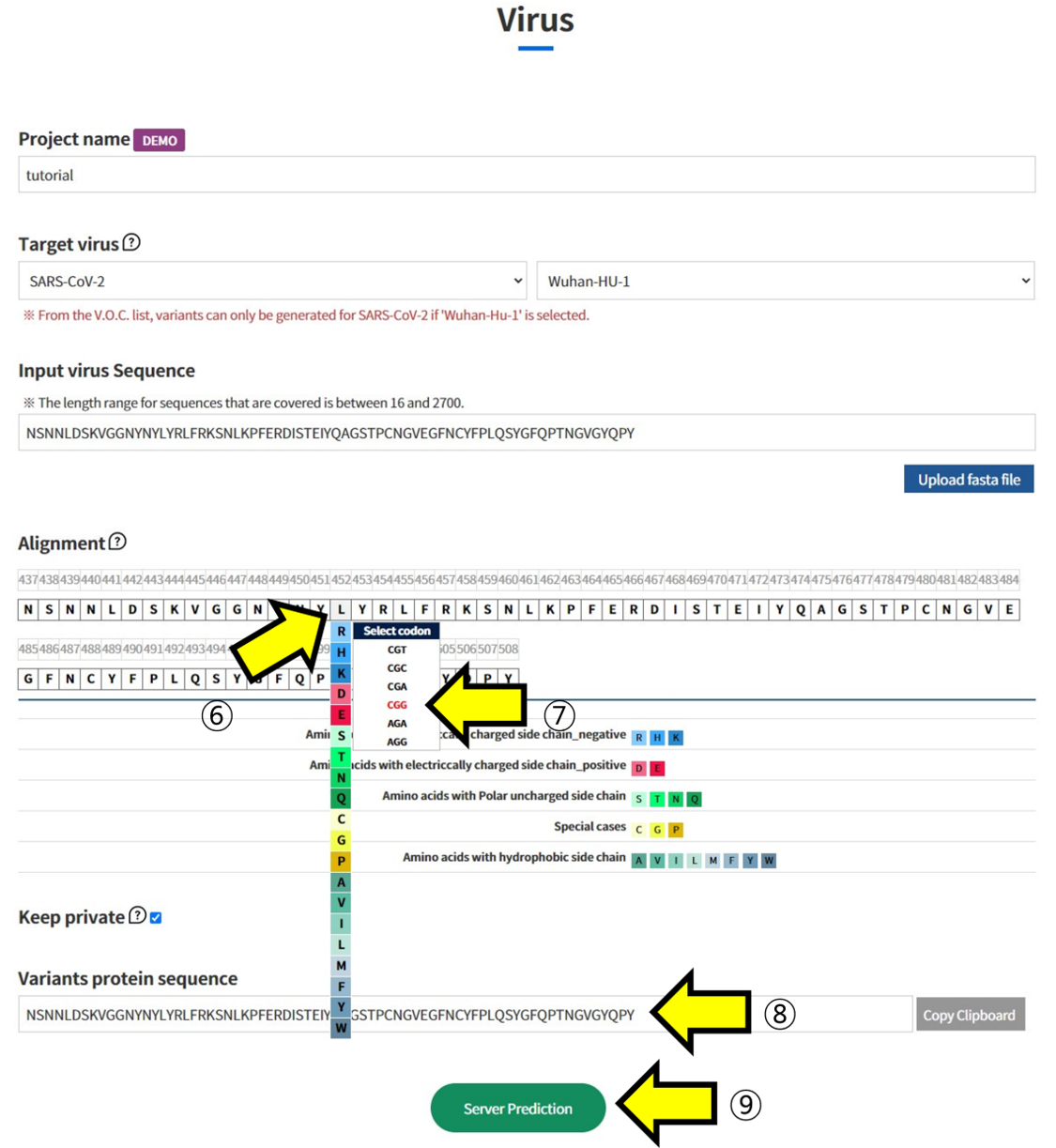
- ⑥ In Alignment, click on the positions in the sequence of Wuhan-HU-1 to select the mutated Amino Acid. ⑦ Additionally, choose which codon to mutate into from the selected Amino Acid. This function is used to assess the impact of mutations at the codon level. ⑧ The sequence with the selected mutations is displayed. ⑨ Submit the task to proceed with the structural prediction and analysis of the mutated sequence.
User sequence - Monomer
- ① Select "All viruses" in the Target Virus section. ② Enter the Amino Acid sequence you want to check in the "Input virus Sequence" box. ③ Alternatively, you can upload a fasta file instead of entering it directly. ④ When the upload window appears, select the fasta file you want to check to pull up the sequence.
User sequence
- ⑥ The Amino Acid sequence recorded in the fasta file you uploaded is displayed in “Alignment”. ⑦ Click on the positions in the displayed sequence where you want to introduce mutations and select the mutated Amino Acid. ⑧ Additionally, choose which codon to mutate into from the selected Amino Acid. This function is used to analyze the impact of mutations at the codon level. ⑨ The sequence with the selected mutations is displayed. ⑩ Submit the task to proceed with the structural prediction and analysis of the mutated sequence.
User sequence - Multimer
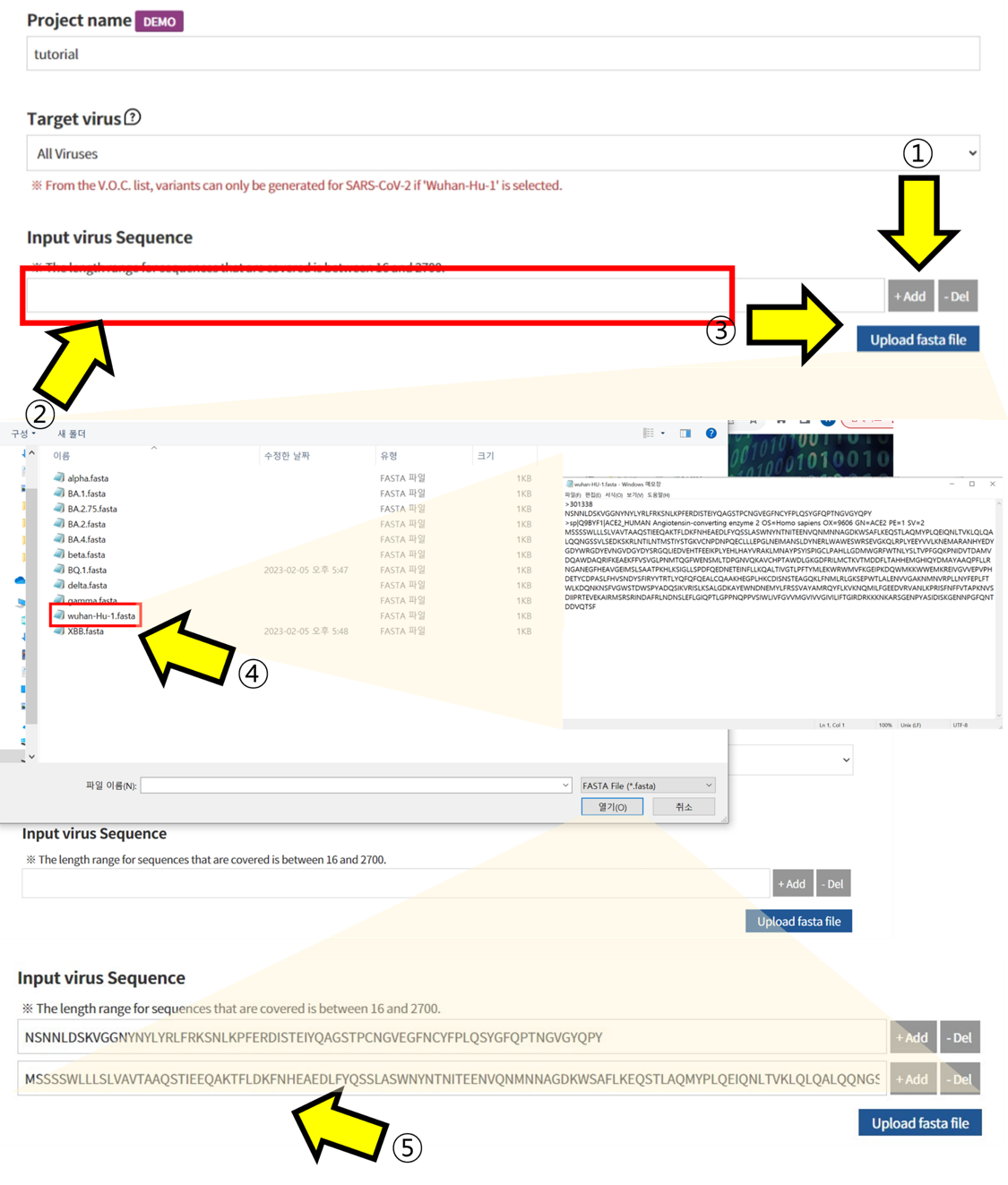
- ① Click the +Add button to create as many sequence input boxes as there are chains in the protein complex you want to predict. ② Enter the Amino Acid sequence of each chain in the generated “Input virus Sequence” boxes. ③ Alternatively, a fasta file can be uploaded without entering the sequence directly ④ When the upload window appears, select the fasta file to retrieve the sequence. ⑤ The sequences of each chain, as stored in the fasta file, are inputted.
User sequence - Multimer

- ⑥ Click on the positions in the displayed sequences where you want to introduce mutations and select the mutated Amino Acid. ⑦ Additionally, choose which codon to mutate into from the selected Amino Acid. This function is used to analyze the impact of mutations at the codon level. ⑧ The sequence with the selected mutations is displayed. ⑨ Submit the task to proceed with the structural prediction and analysis of the mutated sequence.
Result report page - Structure
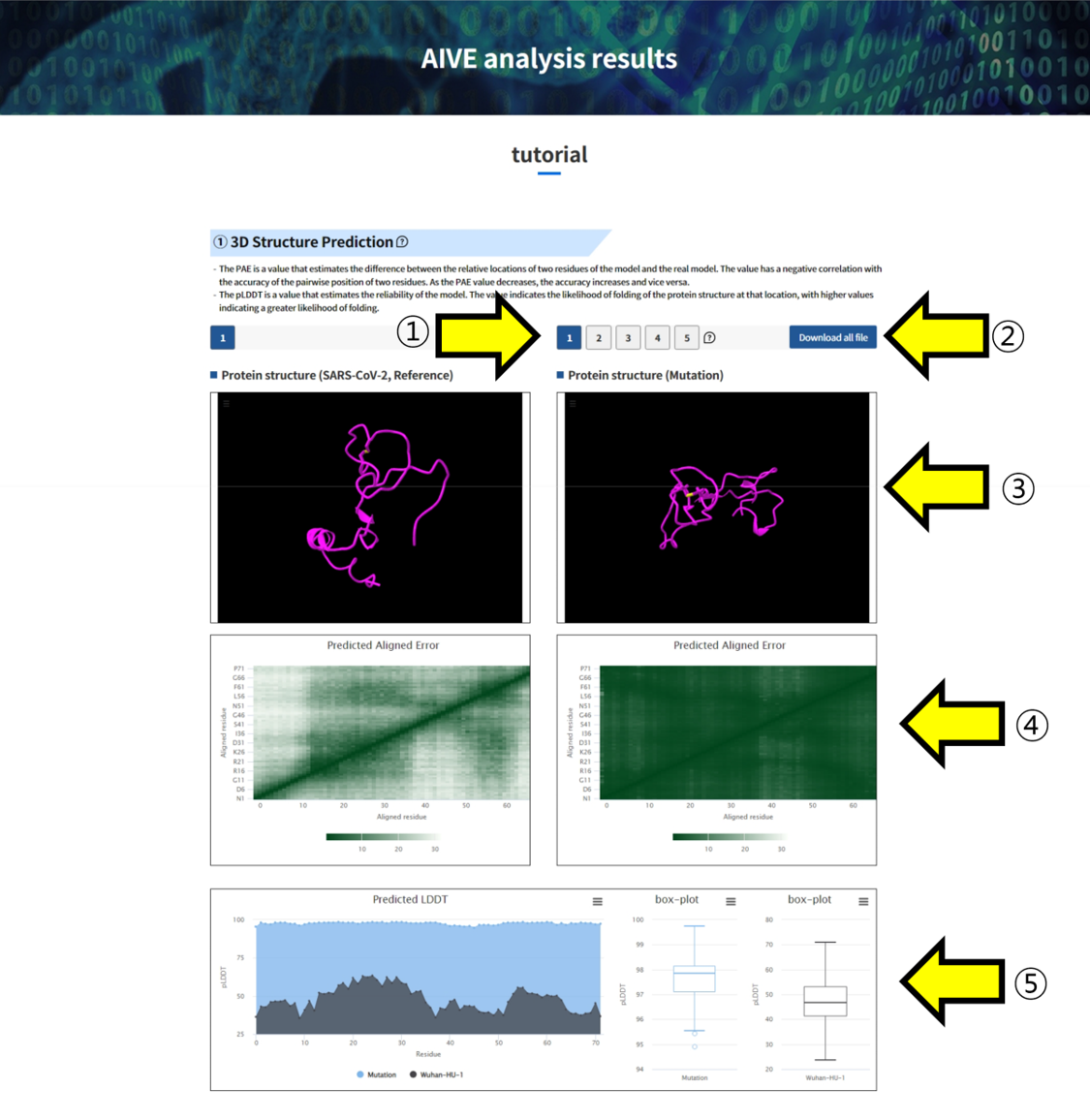
- ① You can select and view the predicted 3D structures by choosing from the five available options. ② Use the "Download all file" button to download the result files of the predicted structures to your device. ③ You can visualize the predicted 3D structure for inspection and comparison with SARS-CoV-2 Wuhan-HU-1. ④, ⑤ Clicking on the highlighted regions in positions allows you to inspect them. ④ Predicted aligned error (PAE) is a value that estimates the difference between the relative locations of two residues of the model and the real model. A low PAE value indicates that the accuracy of the relative location of the two residues is high. • The color at (x, y) indicates AlphaFold’s expected position error at residue x if the predicted and true structures were aligned on residue y. • If the PAE is generally low for residue pairs x, y from two different domains, it indicates that AlphaFold predicts well-defined relative positions and orientations for them. (Explanation from AlphaFold FAQ) ⑤ Predicted LDDT(pLDDT) is a value that estimates the reliability of the model. It estimates how well the actual model residue and predicted model residue match. At the same time, it indicates how well the protein structure folds in the corresponding location. A low pLDDT value indicates that the reliability of the corresponding position is low and that it possesses a disordered structure.
Result report page - APESS
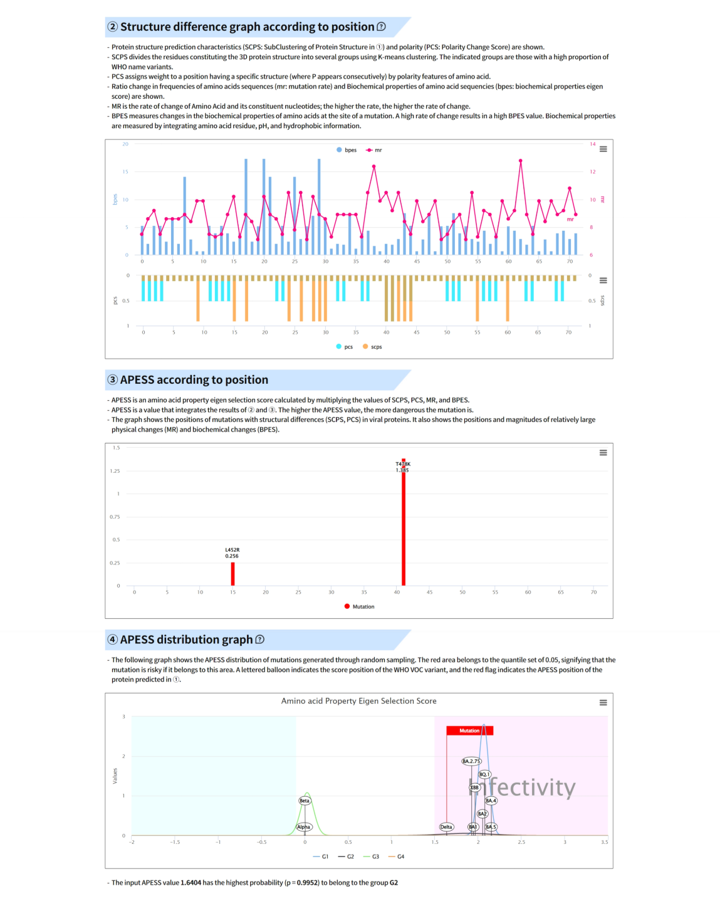
Result report page – APESS subscore

① SCPS
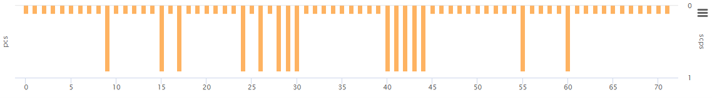
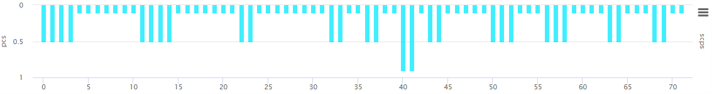
② PCS
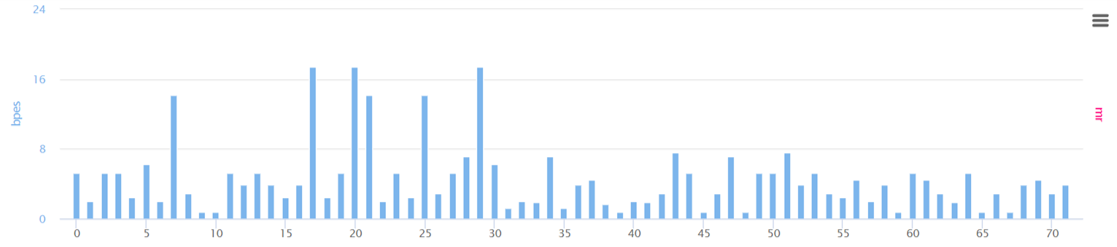
③ BPES
④ MR
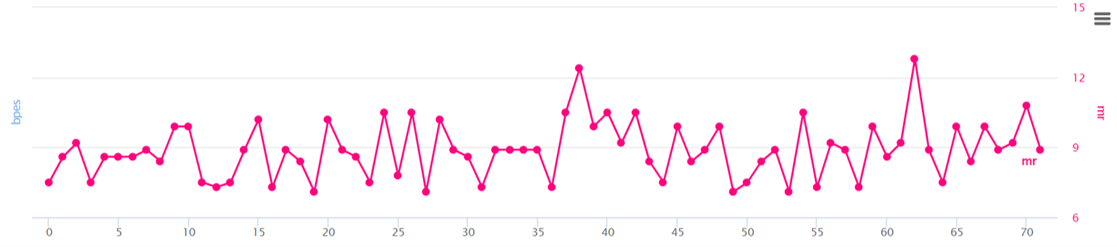
- ① AIVE predicts protein structures due to mutations in each gene for coronavirus lineages or sub-lineages. From the predicted result, it carries out grouping of amino acids (components of 3D protein structure) utilizing K-means clustering to report SCPS results. ② through repeated pattern analysis of polar amino acids in amino acid sequences, AIVE reports PCS results. ③ AIVE figures out amino acid properties to measure BPES through measurement of changes in biochemical properties of amino acid. ④ AIVE calculates MR through rate of change for nucleotide frequencies due to mutations.
Result report page – APESS & distribution
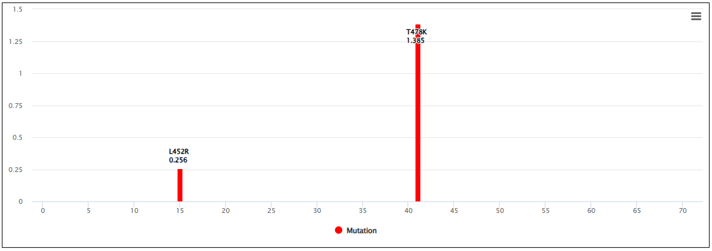
⑤ APESS
⑥ APESS distribution graph
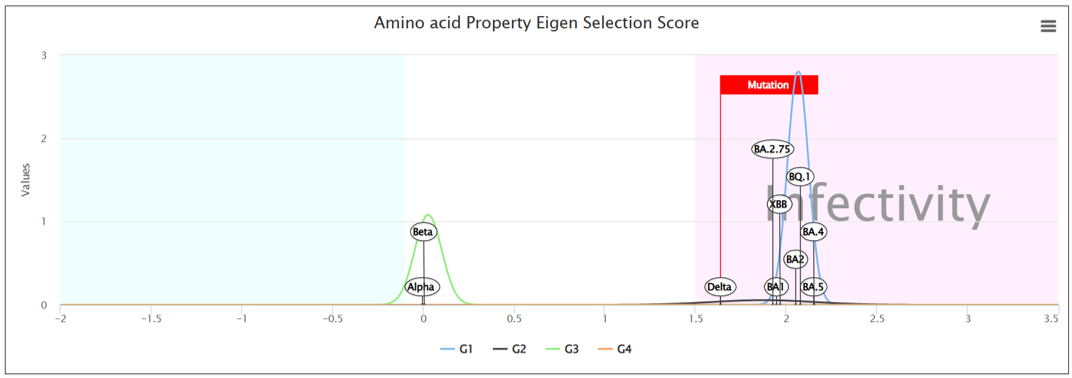
- ⑤ APESS, the result of comprehensive mathematical model (SCP*PCS*MR*BPES) of measured analysis results is provided. ⑥ We calculated the APESS score for a dataset of 7 million samples collected from GISAID. We then applied a Gaussian Mixture Model (GMM) to identify and represent it with 4 components. This allows us to provide the likelihood of which component the sequence is most likely to belong to from the APESS score.
Result report page - Polarity
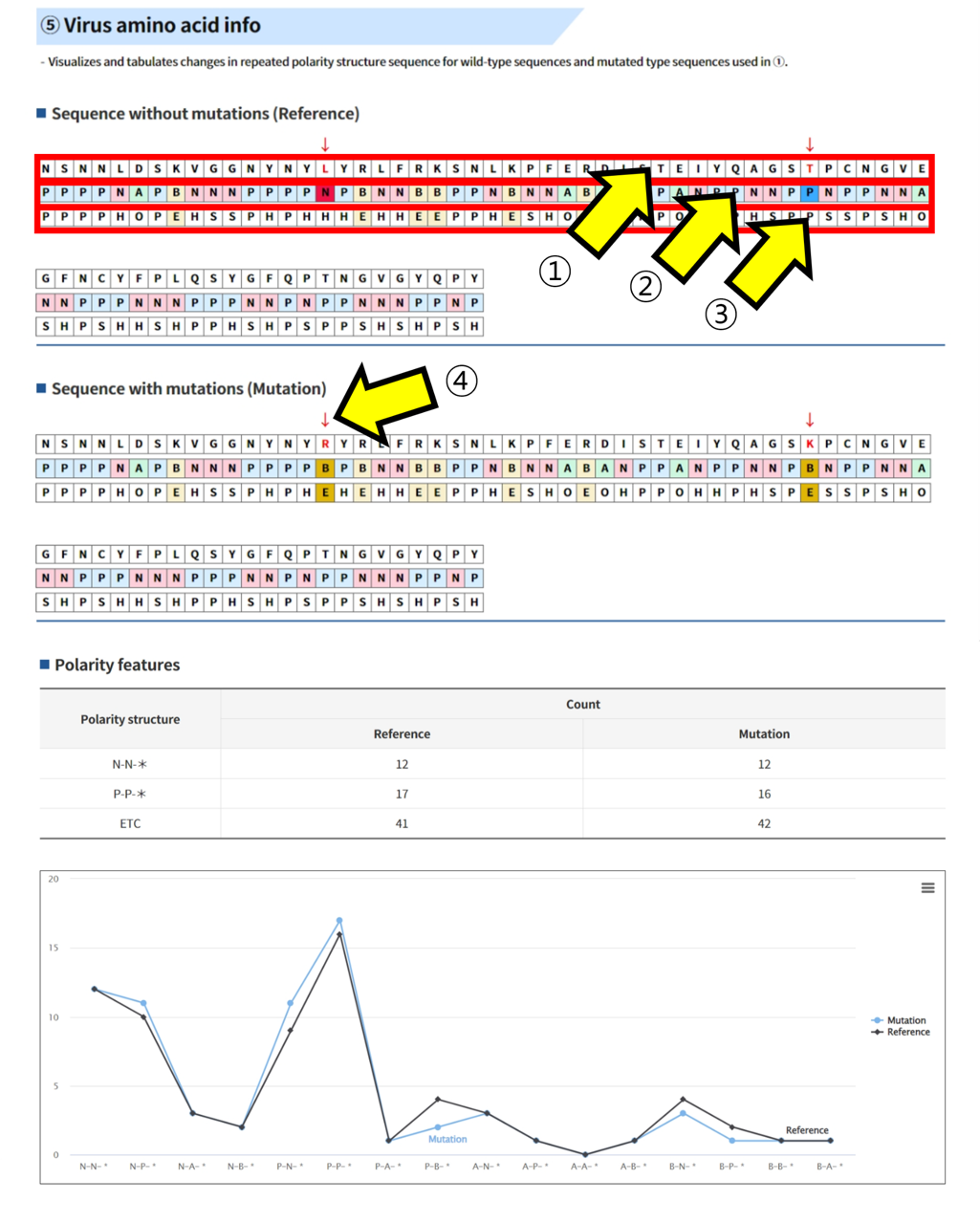
- ① amino acid sequence ② 4 polarity characteristics ③ 5 amino acid properties ④ Mutated positions are indicated in red.
Result report page – All viruses
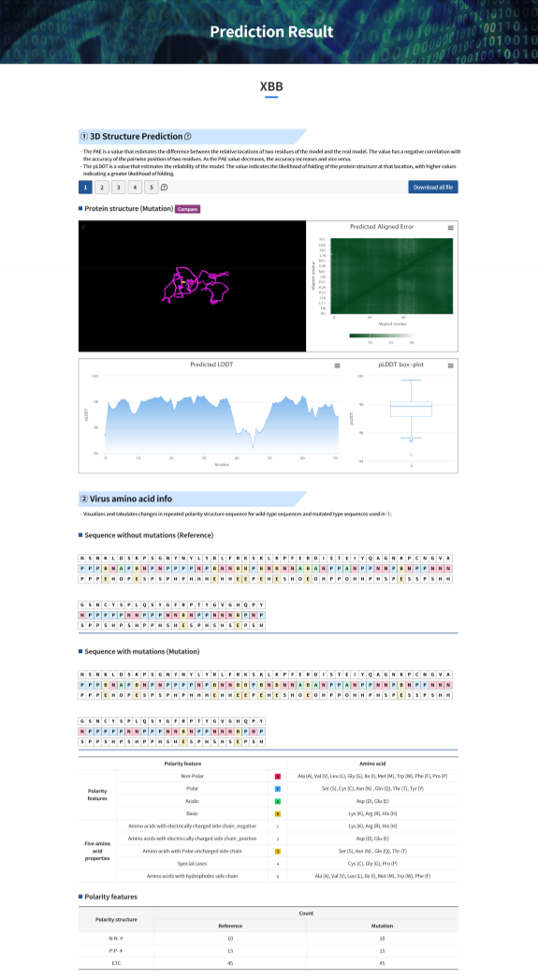
Result report page –Compare
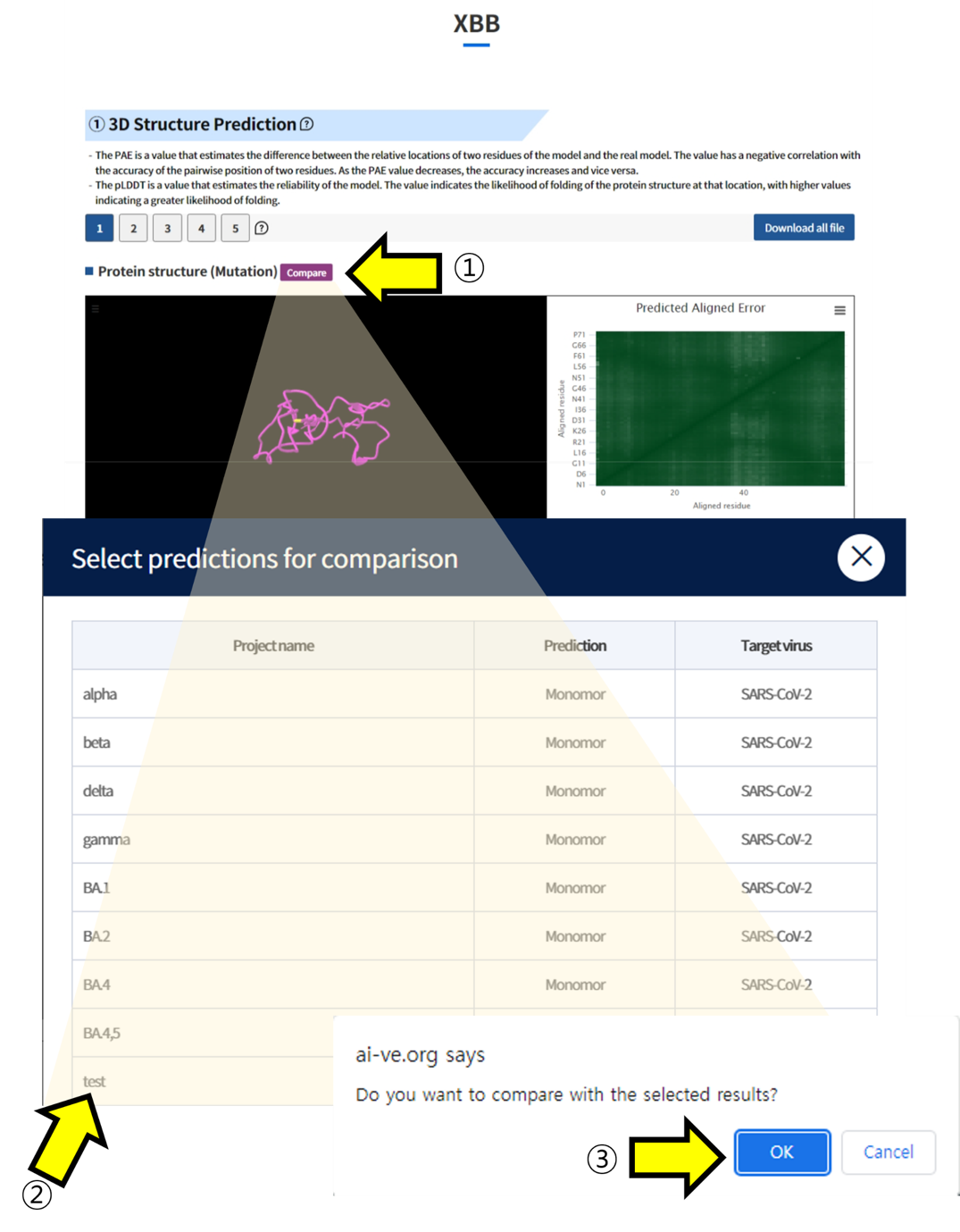
- ① Click the "Compare" button to load the list of other tasks submitted by the user. ② Select the task you want to compare and click on it. ③ Click the "Confirm" button to navigate to the comparison page.
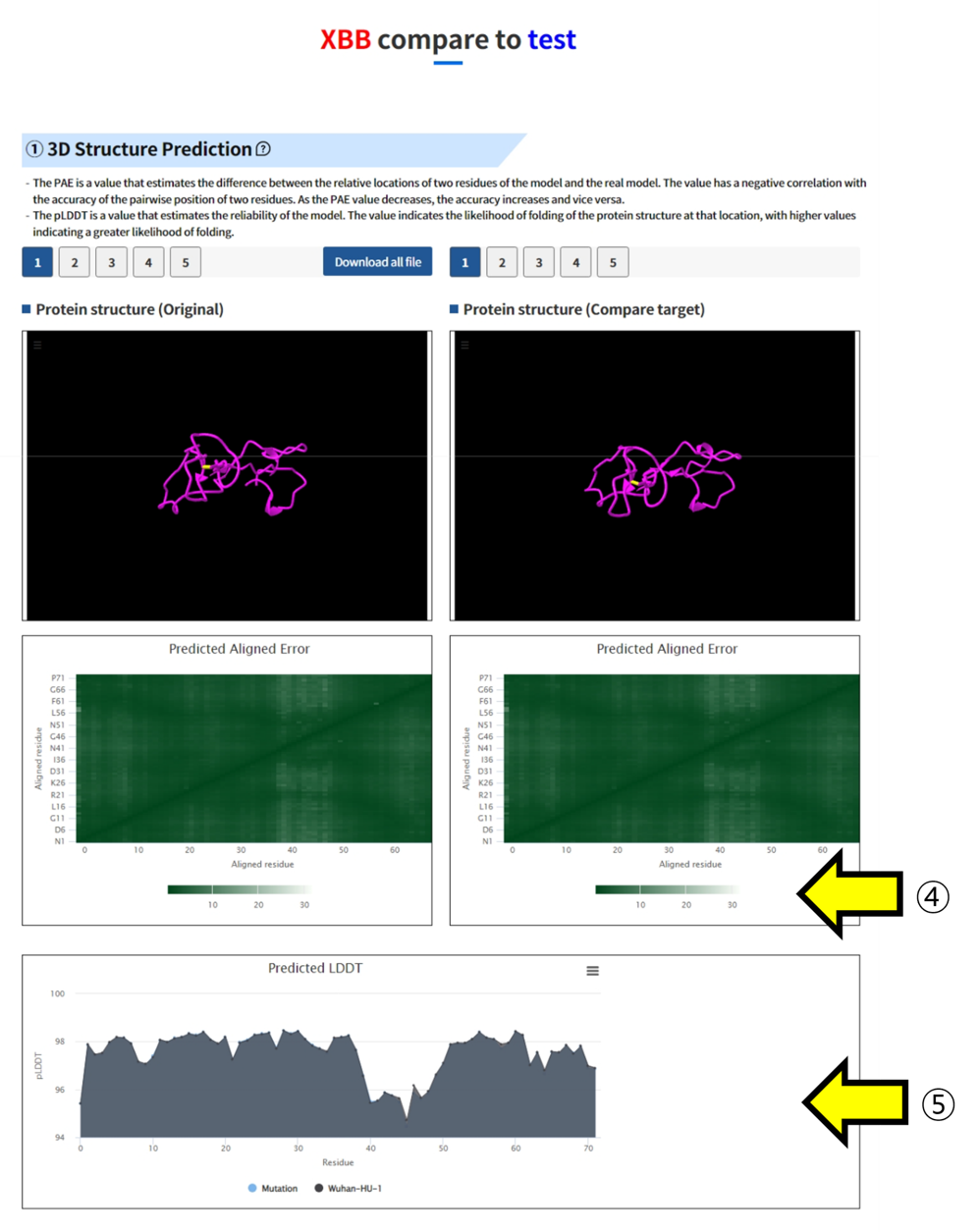
- ④ You can visually inspect the PAE (Predicted Alignment Error) of the two structures using a plot. ⑤ You can compare the pLDDT values of the two structures.
- ⑥ The sequence and polar structures of the two structures can be compared and analyzed.